2 new Exploris 480s arrived
Categories
2 new Exploris 480s arrived


Publication by Thierry Schmidlin in Cell Systems

Paper in Nature Communications

Publication in Science

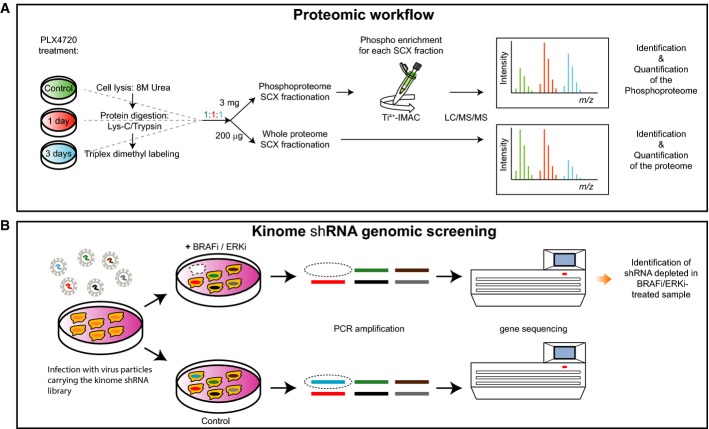
Treatment of BRAF mutant melanomas with specific BRAF inhibitors leads to tumor remission. However, most patients eventually relapse due to drug resistance. Therefore, we designed an integrated strategy using (phospho)proteomic and functional genomic platforms to identify drug targets whose inhibition sensitizes melanoma cells to BRAF inhibition. We found many proteins to be induced upon PLX4720 (BRAF inhibitor) treatment that are known to be involved in BRAF inhibitor resistance, including FOXD3 and ErbB3. Several proteins were down-regulated, including Rnd3, a negative regulator of ROCK1 kinase. For our genomic approach, we performed two parallel shRNA screens using a kinome library to identify genes whose inhibition sensitizes to BRAF or ERK inhibitor treatment. By integrating our functional genomic and (phospho)proteomic data, we identified ROCK1 as a potential drug target for BRAF mutant melanoma. ROCK1 silencing increased melanoma cell elimination when combined with BRAF or ERK inhibitor treatment. Translating this to a preclinical setting, a ROCK inhibitor showed augmented melanoma cell death upon BRAF or ERK inhibition in vitro. These data merit exploration of ROCK1 as a target in combination with current BRAF mutant melanoma therapies.
Smit MA1, Maddalo G2, Greig K1, Raaijmakers LM3, Possik PA1, van Breukelen B3, Cappadona S3, Heck AJ3, Altelaar AF4, Peeper DS5.
Mol Syst Biol. 2014 Dec 23;10:772. doi: 10.15252/msb.20145450
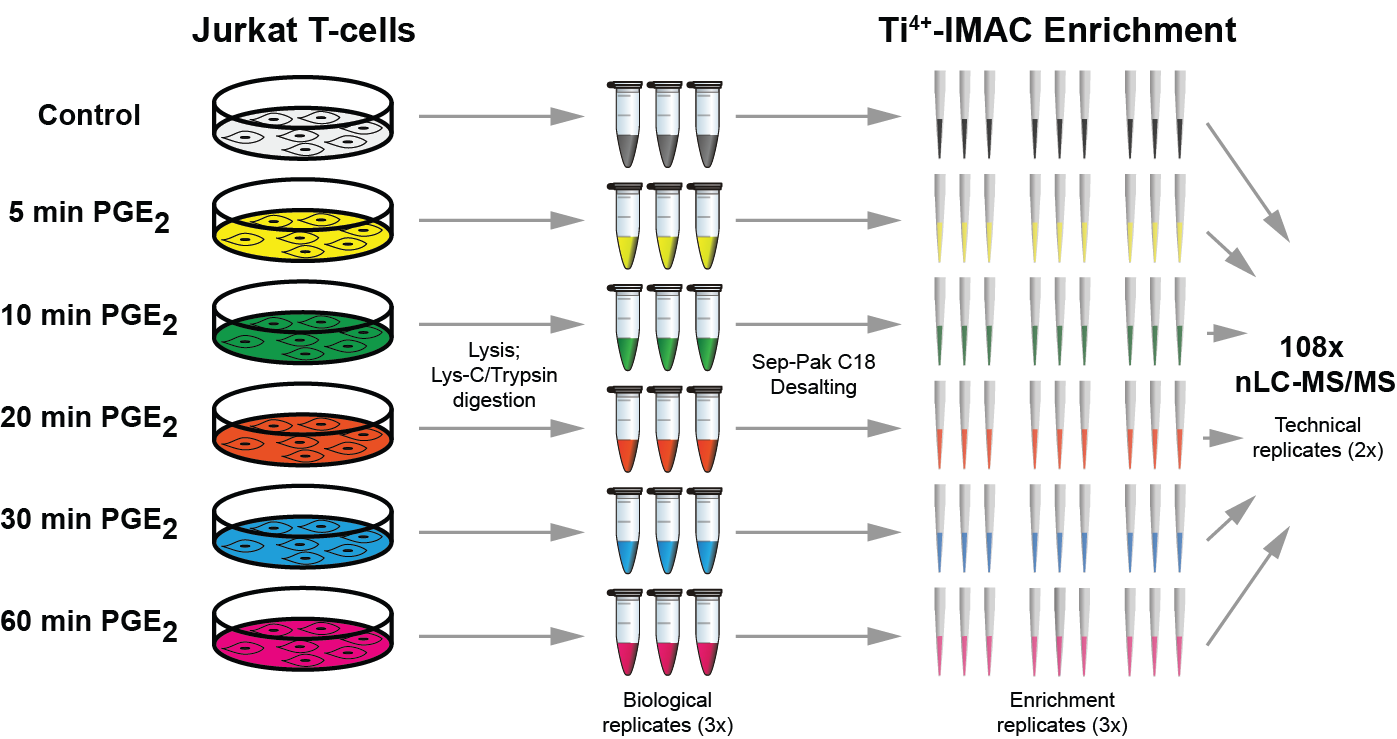
Quantitative phosphoproteomics workflows traditionally involve additional sample labeling and fractionation steps for accurate and in-depth analysis. Here we report a high-throughput, straightforward, and comprehensive label-free phosphoproteomics approach using the highly selective, reproducible, and sensitive Ti(4+)-IMAC phosphopeptide enrichment method. We demonstrate the applicability of this approach by monitoring the phosphoproteome dynamics of Jurkat T cells stimulated by prostaglandin E2 (PGE2) over six different time points, measuring in total 108 snapshots of the phosphoproteome. In total, we quantitatively monitored 12,799 unique phosphosites over all time points with very high quantitative reproducibility (average r > 0.9 over 100 measurements and a median cv < 0.2). PGE2 is known to increase cellular cAMP levels, thereby activating PKA. The in-depth analysis revealed temporal regulation of a wide variety of phosphosites associated not only with PKA, but also with a variety of other classes of kinases. Following PGE2 stimulation, several pathways became only transiently activated, revealing that in-depth dynamic profiling requires techniques with high temporal resolution. Moreover, the large publicly available dataset provides a valuable resource for downstream PGE2 signaling dynamics in T cells, and cAMP-mediated signaling in particular. More generally, our method enables in-depth, quantitative, high-throughput phosphoproteome screening on any system, requiring very little sample, sample preparation, and analysis time.
de Graaf EL, Giansanti P, Altelaar AF, Heck AJ.
Mol Cell Proteomics. 2014 Sep;13(9):2426-2434.
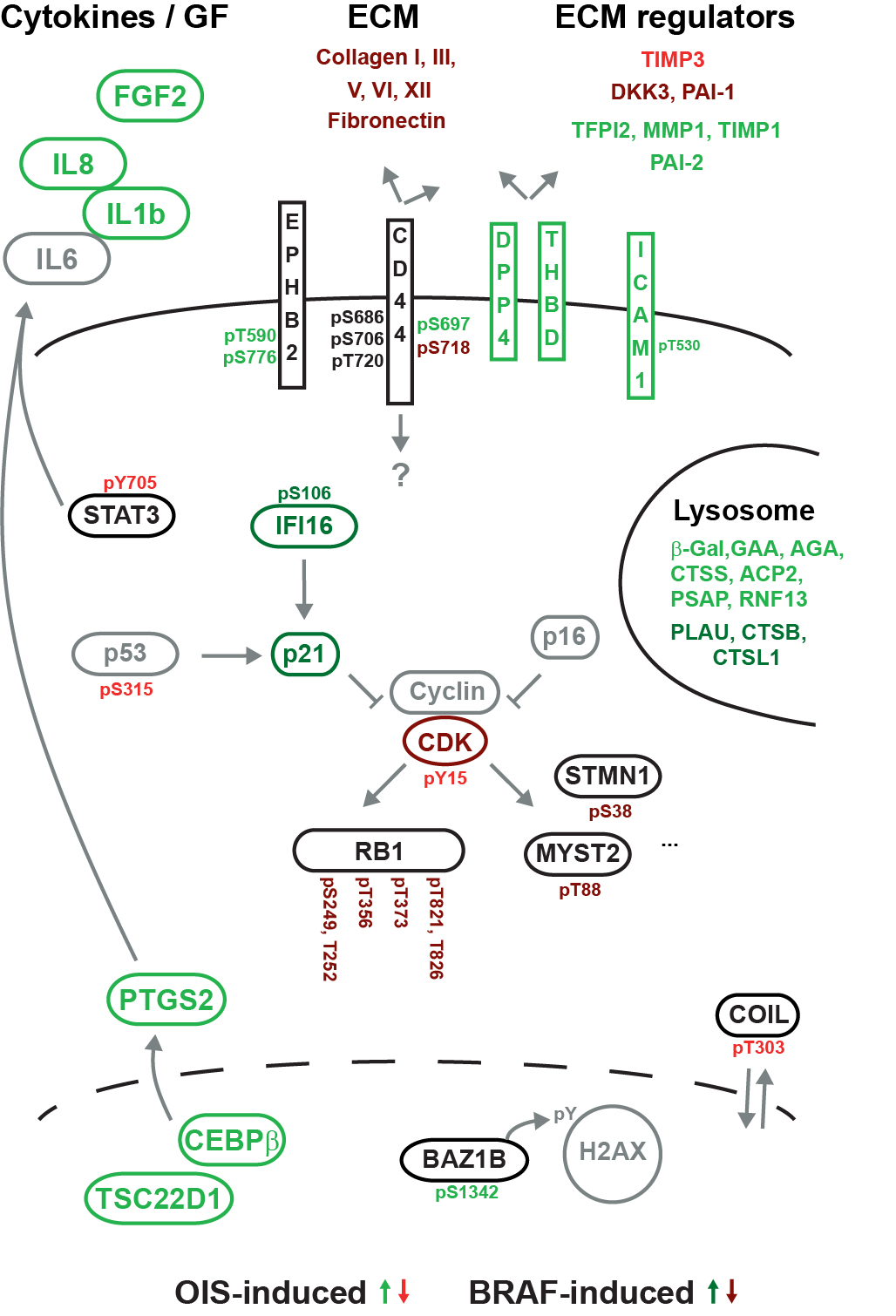
Expression of the BRAF(V600E) oncoprotein is known to cause benign lesions, such as melanocytic nevi (moles). Despite the oncogenic function of mutant BRAF, these lesions are arrested by a cell-autonomous mechanism called oncogene-induced senescence. Infrequently, nevi can progress to malignant melanoma, through mechanisms that are incompletely understood. To gain more insight into this vital tumor-suppression mechanism, we performed a mass-spectrometry-based screening of the proteome and phosphoproteome in cycling and senescent cells and in cells with abrogated senescence. Proteome analysis of senescent cells revealed the up-regulation of established senescence biomarkers, including specific cytokines, but also several proteins not previously associated with senescence, including extracellular matrix-interacting. Using both general and targeted phosphopeptide enrichment by Ti(4+)-IMAC and phosphotyrosine antibody enrichment, we identified over 15,000 phosphorylation sites. Among the regulated phosphorylation sites we encountered components of the interleukin, BRAF/MAPK, and CDK-retinoblastoma pathways and several other factors. The extensive proteome and phosphoproteome dataset of BRAF(V600E)-expressing senescent cells provides molecular clues as to how oncogene-induced senescence is initiated, maintained, or evaded, serving as a comprehensive proteomic basis for functional validation.
de Graaf EL, Kaplon J, Zhou H, Heck AJ, Peeper DS, Altelaar AF.
Mol Cell Proteomics. 2014 Aug;13(8):2089-2100.
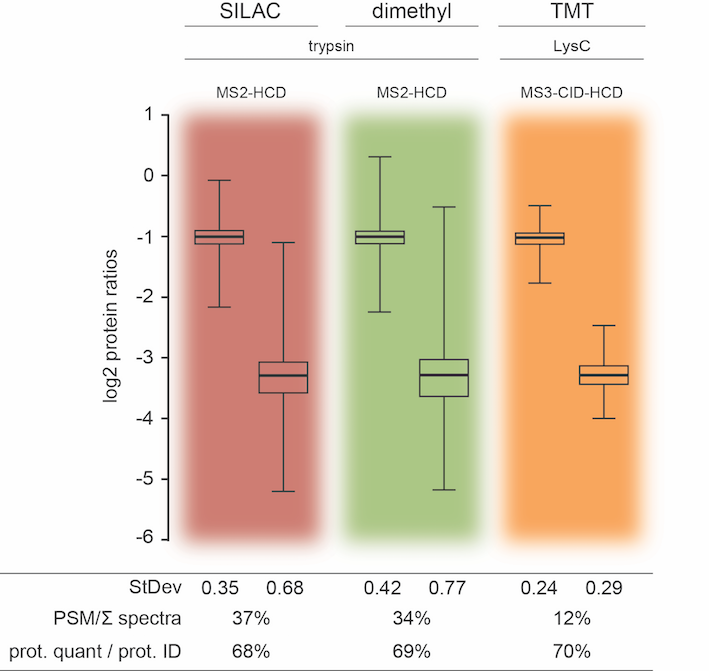
Several quantitative mass spectrometry based technologies have recently evolved to interrogate the complexity, interconnectivity and dynamic nature of proteomes. Currently, the most popular methods use either metabolic or chemical isotope labeling with MS based quantification or chemical labeling using isobaric tags with MS/MS based quantification. Here, we assess the performance of three of the most popular approaches through systematic independent large scale quantitative proteomics experiments, comparing SILAC, dimethyl and TMT labeling strategies. Although all three methods have their strengths and weaknesses, our data indicate that all three can reach a similar depth in number of identified proteins using a classical (MS2 based) shotgun approach. TMT quantification using only MS2 is heavily affected by co-isolation leading to compromised precision and accuracy. This issue may be partly resolved by using an MS3 based acquisition; however, at the cost of a significant reduction in number of proteins quantified. Interestingly, SILAC and chemical labeling with MS based quantification produce almost indistinguishable results, independent of which database search algorithm used.
Altelaar AF, Frese CK, Preisinger C, Hennrich ML, Schram AW, Timmers HT, Heck AJ, Mohammed S.
J Proteomics. 2013 Aug 2;88:14-26.
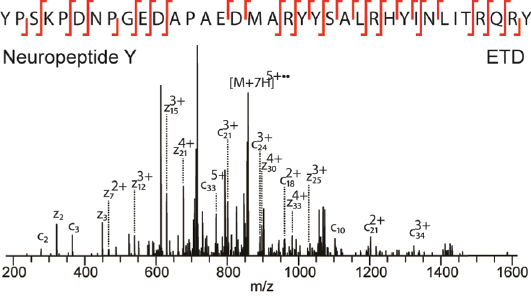
Neuropeptides are intercellular signal transmitters that play key roles in modulation of many behavioral and physiological processes. Neuropeptide signaling in several nuclei in the hypothalamus contributes to the control of food intake. Additionally, food intake regulation involves neuropeptide signaling in the reward circuitry in the striatum. Here, we analyze neuropeptides extracted from hypothalamus and striatum from rats in four differentially treated dietary groups including a high-fat/high-sucrose diet, mimicking diet-induced obesity. We employ high-resolution tandem mass spectrometry using higher-energy collision dissociation and electron transfer dissociation fragmentation for sensitive identification of more than 1700 unique endogenous peptides, including virtually all key neuropeptides known to be involved in food intake regulation. Label-free quantification of differential neuropeptide expression revealed comparable upregulation of orexigenic and anorexigenic neuropeptides in rats that were fed on a high-fat/high-sucrose diet.
Frese CK, Boender AJ, Mohammed S, Heck AJ, Adan RA, Altelaar AF.
Anal Chem. 2013 May 7;85(9):4594-4604.